December 3, 2020
New compact model for gene regulation in higher organisms
Scientists at IST Austria find a way to describe how different molecules interact with each other to activate gene expression.
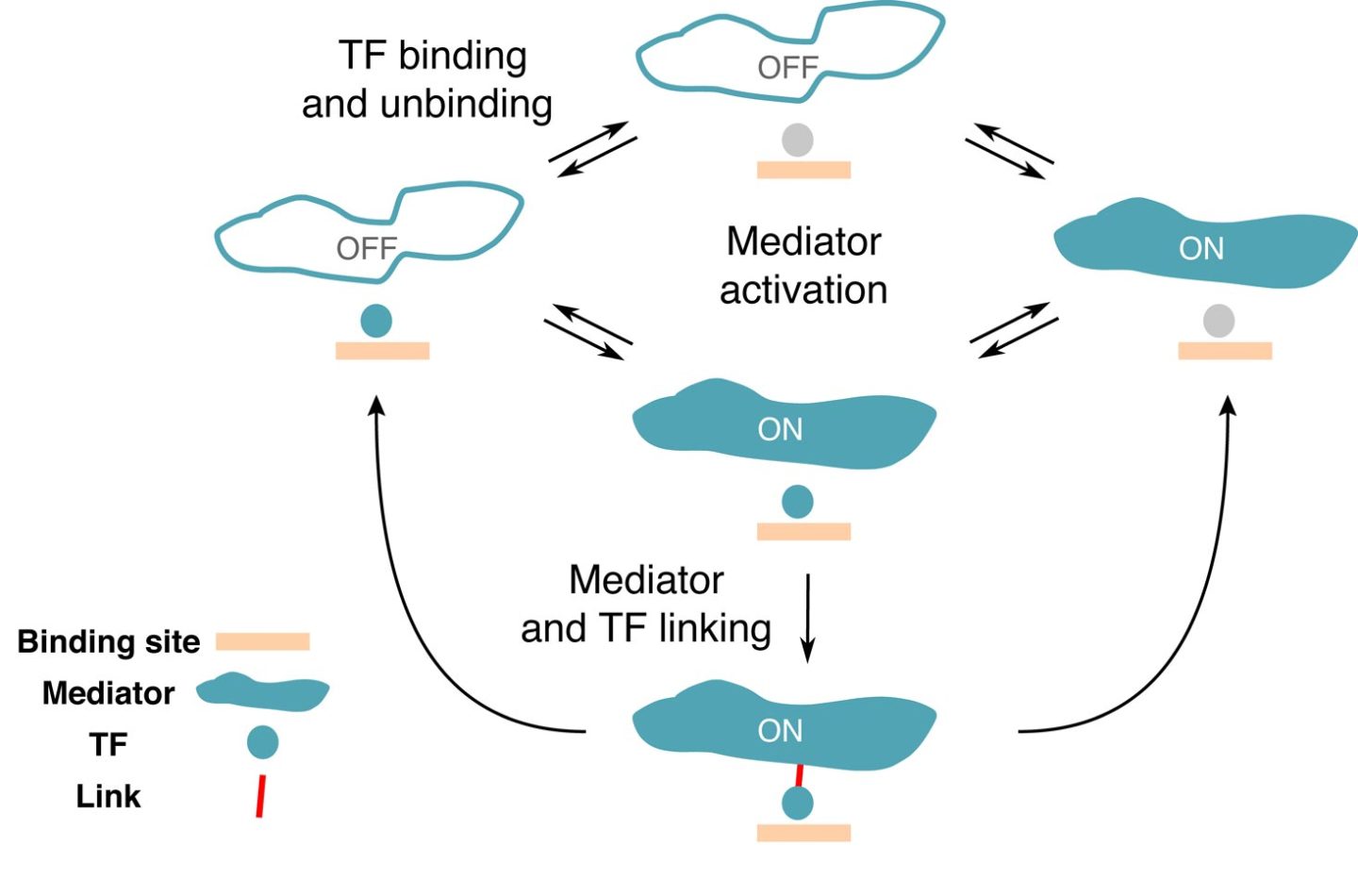
Genes can be turned on and off as needed to adapt to environmental changes. But how do the different molecules involved interact with each other? Scientists from the Institute of Science and Technology Austria (IST Austria) present a candidate mathematical model for gene regulation in eukaryotic cells. The study was published in PNAS.
Although the DNA and its double-helix are one of the most familiar molecules of our time, our knowledge of how cells control what genes they want to express still is rather limited. In order to create, for example, an enzyme, the information that’s inscribed in our DNA about this enzyme needs to be transcribed and translated. To start this highly complex process special regulatory proteins called transcription factors (TFs) bind to specific DNA regions. That way, they can turn the expression of a gene on and off. The big question is: How can transcription factors find the right place on the DNA to properly regulate gene expression?

Simple model – big effect
For prokaryotes – simple cellular organisms without a nucleus, like bacteria – biophysical models already manage to predict gene expression based on the interaction between TFs and DNA regulatory regions. In prokaryotes, the TF binding sites on DNA are rather long and specific, making it easier for the TFs to find their target. In higher organisms called eukaryotes whose cells have a nucleus, mathematically describing the process of gene regulation proved to be much more difficult. Now, a team of researchers at the Institute of Science and Technology Austria (IST Austria) found a way to describe how the interaction between the different regulatory molecules in eukaryotes could look like.

In a new study published in PNAS, Rok Grah, a former graduate student at IST and now a data scientist, working with IST professor Gašper Tkačik and Benjamin Zoller from Princeton University proposed a minimal extension to a classic equilibrium model that can be applied to the switching between the active and inactive states of a gene. To this end, they selected a number of characteristics or “regulatory phenotypes” the desired model should satisfy. “We wanted a gene with a high specificity, meaning that the gene is activated only by the right TFs,” says Rok Grah. Another regulatory phenotype included in the model was the TF residence time on a specific region and a random region of the DNA. “We were able to show that there is a class of simple models that perform well on all of these phenotypes, which wasn’t done so far,” explains Benjamin Zoller. Even though the proposed extension to classical model was minimal, it revealed a wealth of qualitatively new, non-equilibrium behaviors that are consistent with current experimental constraints.
Noisy genes
Based on existing data, the researchers argued that individual TFs are limited in their ability to differentiate between specific and random sites on the DNA. Therefore, although each type of TF preferentially binds certain regulatory DNA sequences, TFs bind other non-cognate targets, too. “The main motivation was to find a model to describe how the regulatory elements on the DNA don’t get activated by non-cognate transcription factors,” says Benjamin Zoller. Their findings suggest that high specificity of gene expression must be a collective effect of the regulatory molecules operating in the “proofreading regime”.

Furthermore, if a gene is active, the number of proteins it produces fluctuates, creating what scientists call gene expression noise. “What surprised me was the tradeoff between noise and specificity. It seems like if you want to have high specificity, it tends to lead to more noise, which is intriguing,” says Benjamin Zoller. High noise is often thought to be detrimental for cells, yet genes in eukaryotes are quite noisy. “So far, we don’t really know why this whole transcription machinery has evolved that way. Perhaps an explanation is that high noise is unavoidable if you want high specificity. Within our model, there seems to be no way around it. High specificity will always mean high noise, and it is possible cells have evolved mechanisms to lower the noise later on in the gene expression process,” adds Rok Grah. The next step in the collaboration is the experimental test of the new model. Its simplicity makes it perfectly suited for confrontation with precise real-time gene expression measurements, for example, on perturbed DNA regulatory sequences.
Publication
Grah R, Zoller B, Tkačik G. 2020. Non-equilibrium models of optimal enhancer function. PNAS. DOI: https://doi.org/10.1073/pnas.2006731117
Funding information
The IST Austria project part was supported by funding from the Human Frontiers Science Program (RGP0034/2018). Rok Grah was supported by the Austrian Academy of Sciences DOC fellowship.
