24. Oktober 2024
Wie Mutationen Krankheiten beeinflussen
ISTA-Forschende präsentieren neuen Ansatz zur Erforschung „polygener“ Krankheiten
Der genetische Code des Menschen ist vollständig kartiert. Dieser Bauplan ermöglicht es Wissenschafter:innen, Genomregionen und deren Variationen zu identifizieren, die für Krankheiten verantwortlich sind. Mit herkömmlichen statistischen Methoden lassen sich diese genetischen „Nadeln im Heuhaufen“ effektiv aufspüren. Komplexer wird es, wenn mehrere Gene zu Krankheiten beitragen, wie es bei Diabetes oder Schizophrenie der Fall ist. Eine neue Studie des Institute of Science and Technology Austria (ISTA) im Journal PNAS befasst sich mit diesem Problem.

Wissenschafter:innen verwenden unzählige statistische Modelle und Algorithmen. Diese kann man sich als eine „Black Box“ vorstellen: leistungsstarke Werkzeuge, die genaue Vorhersagen liefern, mit oftmals unverständlicher und nicht interpretierbarer Funktionsweise. Im Zeitalter des Deep Learning, in dem immer größere Datenmengen verarbeitet werden können, hat Natália Ružičková, Physikerin und Doktorandin am Institute of Science and Technology Austria (ISTA), beschlossen, einen Schritt zurückzumachen. Zumindest im Bereich der Analyse von genetischen Daten.
Gemeinsam mit Michal Hledík, einem ISTA-Absolventen, und Professor Gašper Tkačik schlägt Ružičková nun ein neues statistisches Modell vor. Dieses könnte dabei helfen „polygene Krankheiten“ zu analysieren, bei denen viele Regionen im Genom zu einer Fehlfunktion beitragen, und zu verstehen, warum die identifizierten genomischen Regionen zu diesen Krankheiten beitragen. Dazu kombinierten die Forschenden Genomanalysetechniken mit Erkenntnissen der Grundlagenbiologie. Die Ergebnisse wurden in PNAS veröffentlicht.
Das menschliche Genom entschlüsseln
1990 wurde das Humane Genome Project ins Leben gerufen, um die menschliche DNA – den genetischen Bauplan des Menschen – vollständig zu entschlüsseln. Im Jahr 2003 wurde das Projekt abgeschlossen und ebnete den Weg für zahlreiche Durchbrüche in Wissenschaft, Medizin und Technologie. Durch die Decodierung des menschlichen genetischen Codes erhofften sich die Wissenschafter:innen, mehr über Krankheiten zu erfahren, die mit bestimmten Mutationen und Variationen zusammenhängen. Das menschliche Genom mit seinen 20.000 Genen und noch mehr Basenpaaren – die Buchstaben des Bauplans – erforderte eine große statistische Leistungsfähigkeit. Dies führte zur Entwicklung der so genannten „Genome Wide Association Studies“ (GWAS).
GWAS sind Forschungsansätze zur Ermittlung genetischer Varianten, die potenziell mit Merkmalen eines Organismus wie beispielsweise die Körpergröße, aber auch mit der Neigung zu verschiedenen Krankheiten zusammenhängen. Für letzteres ist das zugrundeliegende statistische Prinzip recht einfach: Die Teilnehmer:innen werden in zwei Gruppen eingeteilt – gesunde und kranke Personen. Ihre DNA wird analysiert, um Variationen – Veränderungen in ihrem Genom – zu entdecken, die bei den von der Krankheit Betroffenen stärker ausgeprägt sind.
Ein Zusammenspiel von Genen
Als diese Korrelationsstudien aufkamen, erwarteten die Wissenschafter:innen, nur einige Mutationen in bekannten Genen zu finden, die mit einer Krankheit in Verbindung stehen und den Unterschied zwischen gesunden und kranken Menschen erklären würden. Die Wahrheit ist jedoch komplizierter. „Manchmal gibt es Hunderte oder Tausende von Mutationen, die mit einer bestimmten Krankheit verbunden sind“, erklärt Ružičková. „Das war überraschend und stand im Widerspruch zu dem, was wir glaubten über die Biologie zu wissen.“
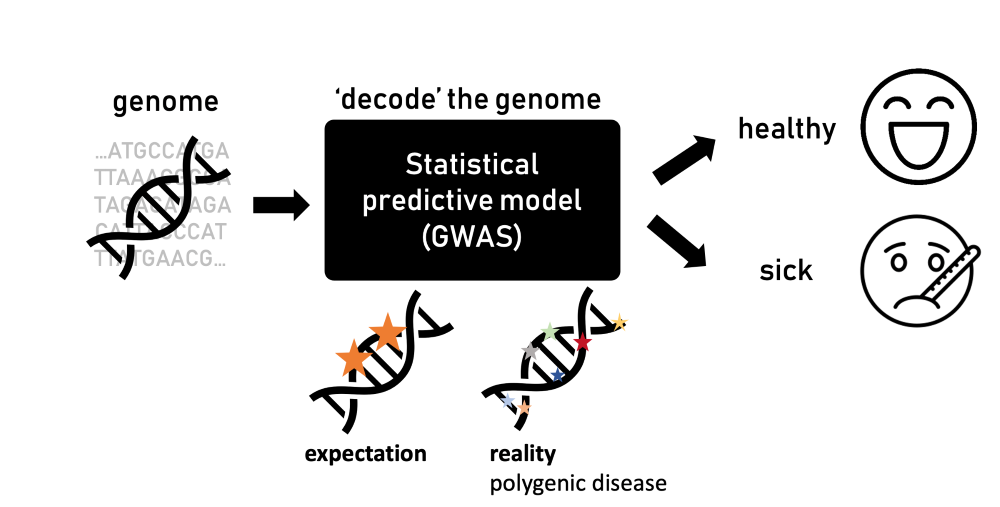
Jede einzelne Mutation hat nur einen minimalen Einfluss auf das Risiko, eine Krankheit zu entwickeln. Zusammen können sie jedoch zum Teil erklären, warum manche Menschen krank werden. Solche Krankheiten werden als „polygen“ bezeichnet. So kann beispielsweise Typ-2-Diabetes nicht auf ein einziges Gen zurückgeführt werden, viel mehr sind es Hunderte Mutationen die daran beteiligt sind. Einige dieser Mutationen wirken sich auf die Insulinproduktion, die Insulinwirkung oder den Glukosestoffwechsel aus und befinden sich meistens in Genomregionen, die bisher nicht mit Diabetes in Verbindung gebracht wurden oder deren biologische Funktionen noch nicht geklärt ist.
Das omnigene Modell
2017 schlugen Evan A. Boyle und Kolleg:innen von der Stanford University 2017 einen neuen konzeptionellen Rahmen vor, das sogenannte „omnigene Modell“. Ihre Erklärung, warum so viele Gene zu Krankheiten beitragen, lautete wie folgt: Zellen verfügen über regulatorische Netzwerke, die Gene mit unterschiedlichen Funktionen verbinden.
„Da Gene miteinander verbunden sind, kann sich eine Mutation in einem Gen auf andere Gene auswirken. Die Wirkung der Mutation kann sich so über das regulatorische Netzwerk ausbreiten“, so Ružičková. Aufgrund dieser Netzwerke tragen letztendlich viele Gene im Regulationssystem zu einer Krankheit bei. Bislang wurde dieses Modell jedoch nicht mathematisch formuliert und blieb eine konzeptionelle Hypothese, die schwer zu testen war. In ihrer aktuellsten Arbeit stellen Ružičková und ihre Kolleg:innen eine neue mathematische Formalisierung namens „quantitatives omnigenes Modell“ (QOM) vor.
Statistik und Biologie kombinieren
Um das Potenzial von QOM zu demonstrieren, mussten die Forschenden das Modell auf ein gut charakterisiertes biologisches System anwenden. Sie entschieden sich für das im Labor übliche Hefemodell Saccharomyces cerevisiae, besser bekannt als Bier- oder Bäckerhefe. Es handelt sich um einen einzelligen Eukaryoten, d. h. seine Zellstruktur ähnelt der von komplexen Organismen wie dem Menschen. „In der Hefe haben wir ein recht gutes Verständnis davon, wie die regulatorischen Netzwerke, die die Gene miteinander verbinden, strukturiert sind“, erklärt Ružičková.
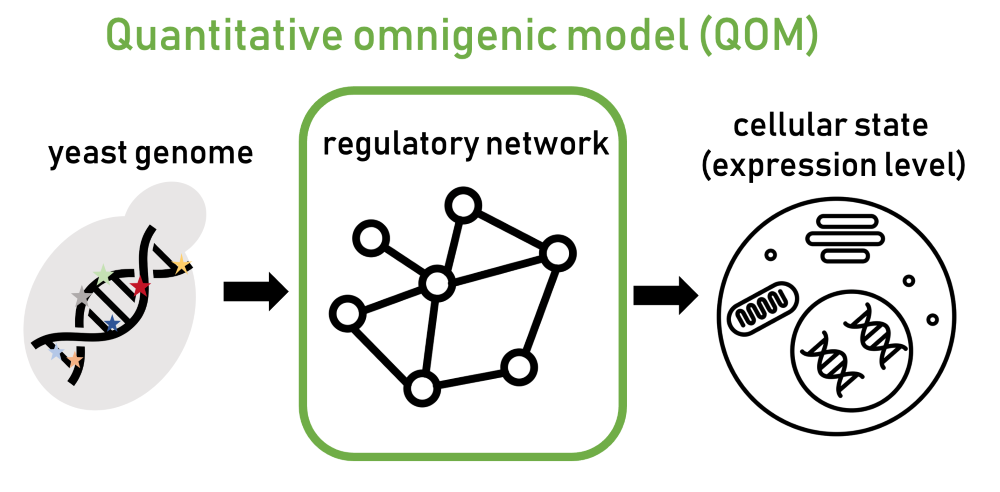
Mithilfe ihres Modells konnten die Wissenschafter:innen zwei Sachen vorhersagen. Erstens, das Ausmaß der Genexpression – die Intensität der Genaktivität, die angibt, wie viel Information der DNA aktiv genutzt wird – und zweitens, wie sich Mutationen im regulatorischen Netzwerk der Hefe ausbreiten. Die Vorhersagen waren äußerst effizient: Das Modell identifizierte nicht nur die relevanten Gene, sondern konnte auch eindeutig feststellen, welche Mutation am wahrscheinlichsten zu einem bestimmten Ergebnis beigetragen hat.
Die Puzzlestücke der polygenen Krankheiten
Das Ziel der Wissenschafter:innen bestand nicht darin, die Standard-GWAS in der Vorhersageleistung zu übertreffen, sondern vielmehr darin, eine andere Richtung einzuschlagen, indem sie das Modell interpretierbar machten. Während ein Standard-GWAS-Modell wie eine „Black Box“ funktioniert und eine statistische Erklärung dafür liefert, wie häufig eine bestimmte Mutation mit einer Krankheit verbunden ist, bietet das neue Modell auch einen kausalen Mechanismus für die Kette von Ereignissen, wie diese Mutation zu einer Krankheit führen kann.
In der Medizin hat das Verständnis der biologischen Zusammenhänge enorme Auswirkungen auf die Suche nach neuen Behandlungsansätzen. Obwohl das Modell derzeit noch weit von einer medizinischen Anwendung entfernt ist, hat es das Potenzial, mehr über polygene Krankheiten preiszugeben. „Wenn man genügend Wissen über die regulatorischen Netzwerke hat, könnte man ähnliche Modelle auch für andere Organismen erstellen. Wir haben uns die Genexpression in Hefe angesehen – ein erster Schritt und Beweis für das Prinzip. Jetzt, wo wir wissen, was möglich ist, kann man über Anwendungen in der Humangenetik nachdenken“, so Ružičková.
Publikation:
Natália Ružičková, Michal Hledík & Gašper Tkačik. 2024. Quantitative omnigenic model discovers interpretable genome-wide associations. PNAS. DOI: 10.1073/pnas.2402340121
Projektförderung:
Dieses Projekt wurde mit Mitteln der Österreichischen Akademie der Wissenschaften durch das DOC-Stipendium 26917 für Natália Ružičková und teilweise durch das Human Frontiers Science Program Grant RGP0034/2018 für Michal Hledík und Gašper Tkačik unterstützt.
